Le syndrome de Crigler-Najjar (CNS) est une pathologie génétique dans laquelle la bilirubine (un pigment produit par le foie lors de la dégradation des globules rouges) n’est pas correctement éliminée par le foie. Le syndrome de Crigler-Najjar est dû à une déficience de l’activité hépatique de la bilirubine glucuronosyltransférase (enzyme normalement responsable de la dégradation de la bilirubine).
Selon le périodique Genetics Home Reference (une publication des instituts nationaux américains de la santé (National Institutes of Health, NIH), ce syndrome est extrêmement rare, puisqu’un seul cas est constaté sur 1 000 000 de naissances (Genetics Home Reference, 2012).
Il existe deux types de syndrome de Crigler-Najjar : le syndrome de type 1 ou type 2. Type 1 (syndrome précoce, CN1) : il s’agit de la forme sévère de cette pathologie. La plupart des patients ne survivent pas au-delà de l’enfance. Type 2 (syndrome tardif, CN2) : cette forme de la maladie est moins sévère. La plupart des patients affectés atteignent l’âge adulte.
Les enzymes sont des protéines essentielles qui jouent le rôle de catalyseur biologique de nombreux processus corporels, tout en permettant les réactions chimiques qui régulent les réactions du métabolisme. Lorsque cette enzyme ne fonctionne pas, la bilirubine s’accumule dans l’organisme au lieu de s’éliminer.
Le syndrome de Crigler-Najjar est une maladie génétique. Si l’un des parents ou grands-parents était affecté par ce syndrome, vous êtes plus susceptible de développer cette condition. La forme sévère du syndrome intervient lorsque le gène défectueux est transmis par les deux parents.
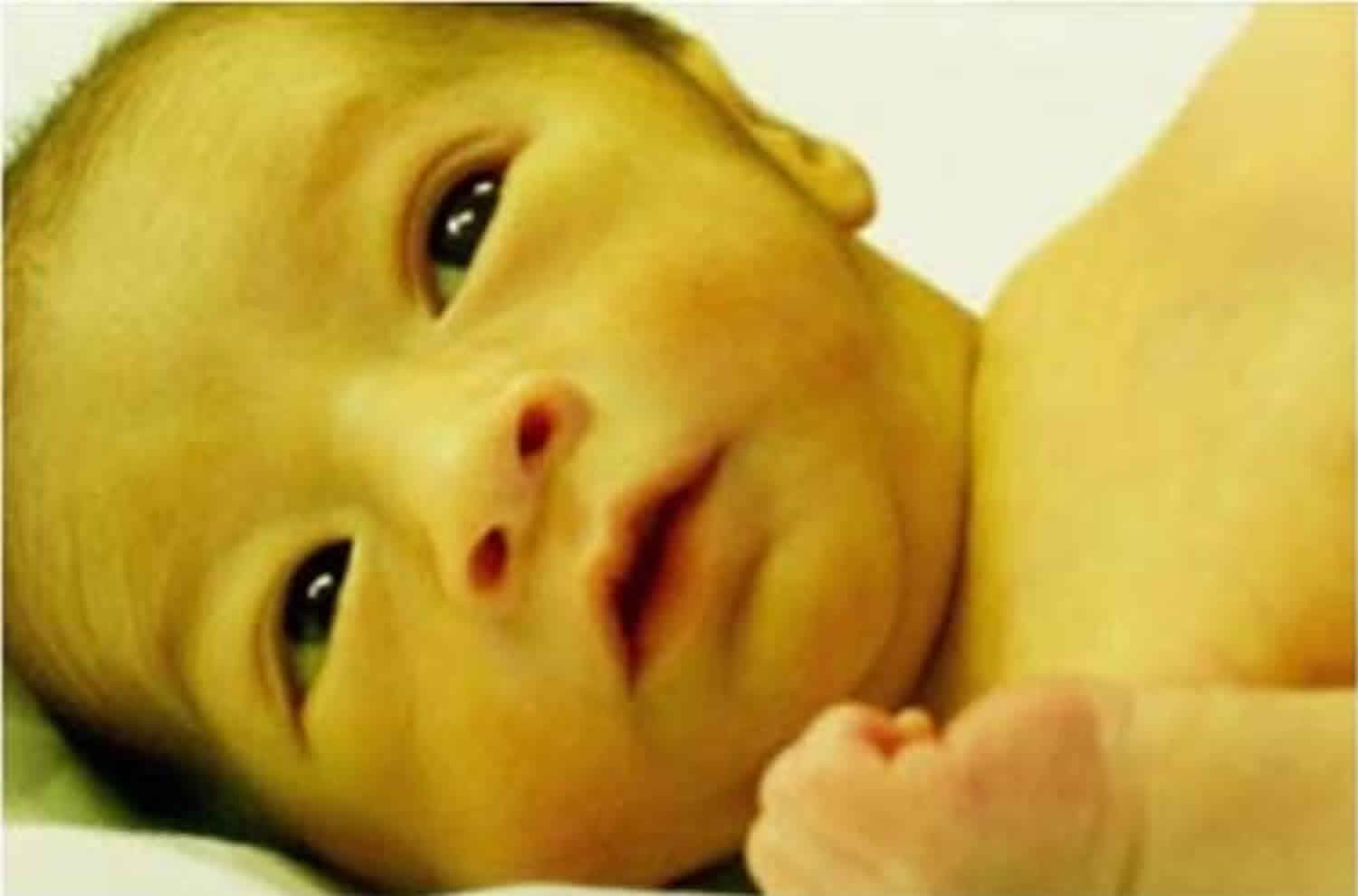
Avec l’accumulation de la biliburine, la peau et les yeux deviennent jaunes, d’où une jaunisse. Ce jaunissement se manifeste généralement à la naissance ou peu après. Il s’agit du signe le plus visible et le plus frappant du syndrome de Crigler-Najjar. Le jaunissement devient généralement plus prononcé avec l’augmentation du taux de biliburine.
En plus de la couleur jaunâtre de la peau et des yeux, un bébé atteint de jaunisse présente éventuellement les symptômes suivants :
- difficulté à se réveiller ;
- difficulté à s’alimenter (inappétence) ;
- incapacité à dormir ;
- incapacité à uriner ou à aller à la selle ;
En l’absence de traitement, la jaunisse entraîne un ictère nucléaire du nouveau-né. L’ictère nucléaire du nouveau-né est une forme de lésion cérébrale, susceptible de conduire à :
- une infirmité motrice cérébrale athétosique (une forme d’infirmité motrice cérébrale (ou d’insuffisance motrice d’origine cérébrale (IMOC)) impliquant des mouvements involontaires des mains, des bras et des jambes) ;
- une perte auditive ;
- des problèmes de vision ;
- un handicap intellectuel.
Consultez immédiatement un médecin en cas de signes de jaunisse ou d’éventuel syndrome de Crigler-Najjar.
Divers facteurs jouent un rôle dans le cadre du diagnostic du syndrome de Crigler-Najjar. Afin d’établir un diagnostic solide, le médecin va prescrire de multiples tests et poser des questions sur les antécédents familiaux.
Pour déterminer la présence éventuelle de la maladie, le médecin va vous interroger sur vos antécédents médicaux et familiaux, ainsi qu’effectuer une analyse de sang. Un échantillon sanguin pourra être analysé afin de détecter la présence de biliburine. Le dosage de la biliburine dans le sang sert à déterminer la présence éventuelle de la maladie.
Chez les nourrissons, le dosage initial de la biliburine est administré à l’aide d’un posemètre. En tenant le posemètre en direction de la tête de l’enfant, l’infirmière/infirmier est en mesure d’évaluer le taux de biliburine présent dans la peau. La présence d’un taux élevé de biliburine signale généralement une jaunisse. En cas de taux élevé, le médecin prescrit généralement une analyse de sang.
Autres examens susceptibles d’être utilisés pour mesurer la biliburine ou la fonction hépatique (fonctionnement du foie) : biopsie du foie (prélèvement d’un petit morceau du foie qui est supprimé et examiné au microscope) et dosage enzymatique (mesure de l’activité de l’enzyme).
Plusieurs types de traitement sont possibles pour le syndrome de Crigler-Najjar. Les principaux traitements sont présentés dans les rubriques suivantes.
Photothérapie.
Le traitement le plus courant pour les bébés atteints du syndrome de Crigler-Najjar est la thérapie par la lumière, ou photothérapie . Cette procédure non invasive expose la peau de l’enfant à une lumière bleue qui va prévenir ou traiter le jaunissement de la peau. Ce traitement est plus efficace chez les enfants, leur peau étant nettement plus fine. Les peaux fines facilitent l’absorption de la lumière. Les adultes ont une peau plus épaisse qui n’absorbe pas facilement la lumière.
Transfusion sanguine
Dans les cas graves, une transfusion sanguine pourra s’avérer nécessaire afin d’éliminer la biliburine du flux sanguin. La transfusion sanguine retire une certaine quantité de sang par un tube, tout en administrant du sang frais dans le corps par un autre tube. En règle générale, cette opération est effectuée en soins externes. Elle prend une à deux heures.
Greffe de foie
Les personnes affectées d’un syndrome de Crigler-Najjar précoce (type 1) peuvent être traitée avec succès par une greffe de foie. Cette solution ne fonctionne pas chez les personnes présentant un syndrome de Crigler-Najjar tardif.
Phénobarbital
Les personnes affectées par le syndrome de Crigler-Najjar de type 2 (tardif) peuvent être traitées à l’aide d’un médicament : le phénobarbital. Ce médicament barbiturique est ordinairement prescrit à des personnes souffrant de convulsions (dont l’épilepsie), mais il est également prescrit pour traiter les symptômes du syndrome de Crigler-Najjar. Ce médicament produit une accoutumance : il est donc prescrit sous étroite surveillance médicale.
Les patients atteints du type 2 (tardif) de la maladie présentent un meilleur pronostic. Le CN2 n’implique pas de lésions organiques majeures. Cependant, ils sont susceptibles de développer une jaunisse.
Les enfants atteints du CN1 exigent un traitement à vie. S’ils survivent, les enfants atteints de cette forme du syndrome présentent généralement des lésions cérébrales majeures à l’âge adulte.